When you take a pill once a day instead of three times, it’s not magic-it’s science. Modified-release (MR) formulations are engineered to control how and when a drug enters your bloodstream. This isn’t just about convenience. For drugs with narrow therapeutic windows-like warfarin or lithium-even small changes in how quickly the drug is absorbed can mean the difference between effective treatment and dangerous side effects. That’s why bioequivalence for these products isn’t the same as for regular pills. The rules are stricter, the testing is more complex, and the stakes are higher.
Why Modified-Release Formulations Need Special Rules
| Parameter | Immediate-Release (IR) | Modified-Release (MR) |
|---|---|---|
| Primary PK Metrics | AUC0-t, Cmax | AUC0-t, Cmax, partial AUC (pAUC) |
| Study Design | Single-dose, fasting | Single-dose preferred; steady-state sometimes required |
| Dissolution Testing | One pH condition | Three pH levels (1.2, 4.5, 6.8) |
| Alcohol Testing | Not required | Required if ≥250 mg active ingredient |
| Acceptance Range | 80.00-125.00% | 80.00-125.00% (or tighter for NTI drugs) |
| Biowaiver Eligibility | Common for BCS Class I | Very limited; requires strong dissolution similarity |
Regular immediate-release tablets dissolve fast and release their entire dose within minutes. Their bioequivalence is straightforward: if the generic matches the brand’s absorption speed and total amount absorbed, it’s considered equivalent. But with modified-release products-like extended-release (ER) tablets, delayed-release capsules, or multiphasic systems-the drug is released over hours, sometimes in stages. A generic version might release the same total amount over 24 hours, but if it dumps too much too early, or too slowly later on, it won’t work the same way.
Take zolpidem tartrate extended-release (Ambien CR). It has two layers: one that releases quickly to help you fall asleep, and another that kicks in later to help you stay asleep. A generic version that only matches the total AUC and Cmax might still fail if it doesn’t replicate the exact timing of those two phases. That’s why regulators now require partial AUC measurements-like AUC from 0 to 1.5 hours and from 1.5 hours to infinity-to prove both parts behave the same.
How Regulatory Agencies Differ
The U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the World Health Organization (WHO) all want the same thing: safe, effective generics. But they don’t always agree on how to prove it.
The FDA leans heavily on single-dose studies. Since 2015, 92% of approved extended-release generics used this approach. Why? Because it’s more sensitive to differences in how the drug is released from the tablet. Multiple-dose studies can mask problems-like inconsistent release patterns-because the drug builds up in the body over time, smoothing out the peaks and valleys.
The EMA, on the other hand, still requires steady-state studies for some MR products, especially when the drug accumulates significantly (accumulation ratio >1.5). This means giving the drug daily for days or weeks until levels stabilize. Critics argue this is outdated and unnecessarily expensive. But supporters say it better reflects real-world use for chronic conditions like hypertension or epilepsy.
And then there’s the WHO, which claims MR bioequivalence rules should be “essentially the same” as for regular drugs. That’s not what the FDA or EMA do. In practice, the WHO’s position is rarely followed by major regulators. It’s a reminder that global harmonization is still a work in progress.
Testing That Goes Beyond Blood Samples
Bioequivalence isn’t just about drawing blood every hour for 24 hours. It’s also about how the drug behaves in the lab before it ever touches a human.
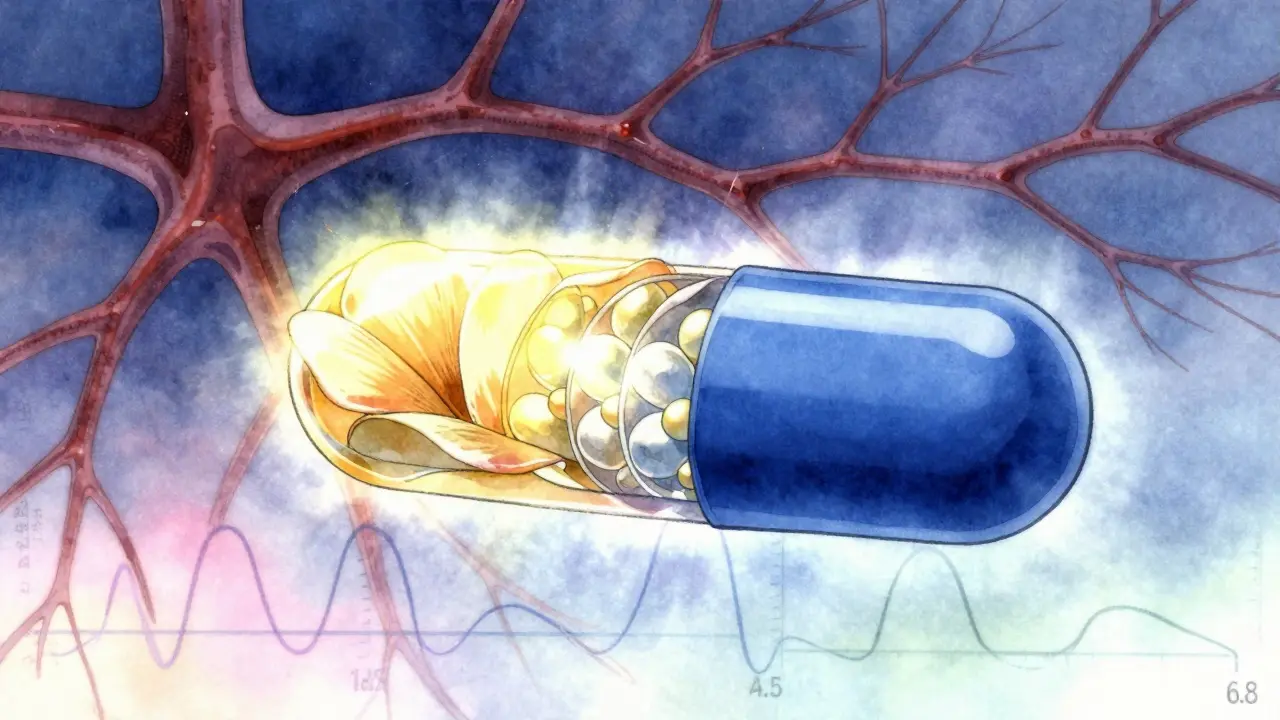
For extended-release tablets, regulators demand dissolution testing at three different pH levels: stomach acid (pH 1.2), small intestine (pH 4.5), and colon (pH 6.8). Why? Because a tablet that works perfectly in a fasting stomach might fall apart too fast in a full one-or not release at all in the colon. The test uses a similarity factor called f2. If the test product’s dissolution profile matches the brand’s with an f2 score of 50 or higher, it’s considered similar. For some products, like beaded capsules, only one pH condition is needed. But for ER tablets? All three.
Then there’s alcohol testing. If a tablet contains 250 mg or more of active ingredient, it must be tested in a solution with 40% alcohol. Why? Because alcohol can cause “dose dumping”-a sudden, dangerous release of the entire drug dose. Between 2005 and 2015, seven ER products were pulled from the market because of this risk. One was an extended-release oxycodone that released 90% of its dose in just two hours when exposed to alcohol.

When the Numbers Aren’t Enough: Highly Variable Drugs
Some drugs, like clopidogrel or carbamazepine, behave wildly differently from person to person. Their absorption varies so much that even a perfect match in average levels might still miss important differences. For these, standard 80-125% bioequivalence limits don’t cut it.
This is where Reference-Scaled Average Bioequivalence (RSABE) comes in. Instead of using fixed limits, regulators scale the acceptance range based on how variable the reference product is. The FDA caps this scaling at 57.38% for the reference’s within-subject variability. That means if the brand drug varies a lot, the generic can vary a bit more too-without compromising safety.
But RSABE isn’t easy. It requires more subjects, longer study durations, and complex statistical models. One formulation scientist told me it adds 6 to 8 months to development time. And if the data isn’t clean? The application gets rejected.
Cost, Complexity, and the High Failure Rate
Developing a generic immediate-release tablet might cost $2-3 million. A modified-release version? $7-9 million. Why? Because the studies are longer, more complex, and harder to pass.
Between 2018 and 2021, 22% of MR generic applications were initially rejected by the FDA-mostly because of incomplete pAUC analysis or failed dissolution profiles. One case that made headlines was the 2012 rejection of a generic version of Concerta (methylphenidate ER). The applicant didn’t properly test the early release phase (0-2 hours), which is critical for ADHD meds that need to work quickly after dosing. The FDA’s complete response letter was blunt: “The proposed product does not demonstrate bioequivalence at the clinically relevant early timepoints.”
Even when companies get it right, it’s expensive. Single-dose MR bioequivalence studies cost $1.2-1.8 million. IR studies? $0.8-1.2 million. And that’s just the clinical part. Add in dissolution method development, statistical modeling, and regulatory writing, and you’re looking at a project that needs a team of 10-15 specialists.

What Happens When Bioequivalence Isn’t Enough?
Here’s the uncomfortable truth: a generic can meet all regulatory bioequivalence criteria-and still not work the same in real life.
A 2016 study in Neurology found that 18% of generic extended-release antiepileptic drugs had higher rates of seizure breakthrough compared to the brand. Patients weren’t failing bioequivalence tests-they were failing in real life. Why? Maybe the generic released the drug a little too slowly at night, or the formulation didn’t handle food the same way. The blood levels looked fine, but the brain didn’t get consistent coverage.
This is why experts like Dr. Donald Mager argue that steady-state studies, while costly, may be necessary for some drugs. And why the FDA is now pushing for more in vitro-in vivo correlation (IVIVC) models. These are mathematical models that link lab dissolution data to actual blood levels. If you can prove your generic’s dissolution profile predicts real-world performance, you might not need as many human studies.
So far, the FDA has accepted Level A IVIVC models for 12 MR products-including extended-release paliperidone. It’s a promising path forward. But it’s not easy. Building a valid IVIVC model takes years of data, advanced software, and deep pharmacokinetic expertise.
What’s Next for Modified-Release Generics?
The market for MR drugs is growing fast. In 2022, it was worth over $325 billion. By 2028, IQVIA predicts it will make up 42% of all prescription sales, driven by aging populations and chronic diseases like diabetes, heart failure, and Parkinson’s.
Regulators are catching up. The EMA’s 2023 draft guideline proposes aligning more closely with the FDA-possibly eliminating the steady-state requirement for most products. The FDA is also working on a new 2024 guidance for complex MR products like gastroretentive systems and multiparticulate capsules.
Meanwhile, companies are turning to mechanistic modeling-like PBPK (physiologically based pharmacokinetic) models-to predict how a drug will behave before they even test it in humans. A 2022 DIA survey found 68% of major pharma firms now use these tools for MR development.
The bottom line? Modified-release generics aren’t just “another pill.” They’re precision-engineered systems. Getting them right demands more than just matching blood levels. It requires understanding release kinetics, dissolution behavior, food effects, alcohol interactions, and how the drug behaves over time in real patients. The science is advanced. The regulations are tough. And the margin for error? Slim.
Why can’t a generic modified-release drug just match the brand’s total drug amount in the blood?
Because the timing of release matters. A drug that releases all at once might cause side effects, while one that releases too slowly might not work at all. Modified-release products are designed to maintain steady levels over time. If the generic releases too fast early on or too late later, even if the total amount absorbed is the same, the patient may experience ineffective treatment or adverse effects. That’s why regulators require partial AUC measurements and dissolution profiles across different pH levels.
Are alcohol tests really necessary for all extended-release pills?
Only if the pill contains 250 mg or more of the active ingredient. Alcohol can disrupt the coating or matrix that controls drug release, causing the entire dose to dump into the bloodstream at once. This can lead to overdose, especially with opioids or stimulants. Between 2005 and 2015, seven ER products were withdrawn because of this risk. Testing in 40% ethanol simulates what happens if someone drinks alcohol while taking the medication.
Why do some generic MR drugs still cause seizures even if they pass bioequivalence tests?
Bioequivalence tests measure average blood levels, but they don’t always capture subtle differences in release patterns over time. For antiepileptic drugs, even a 10-15% delay in peak concentration can reduce effectiveness. Some generics may have slightly slower or inconsistent release in the presence of food or due to minor formulation differences. These aren’t enough to fail a standard test-but they’re enough to cause breakthrough seizures in vulnerable patients.
Is it true that developing a generic MR drug costs more than an IR one?
Yes, significantly more. A typical immediate-release generic costs $2-3 million to develop. A modified-release version can cost $7-9 million. That’s because MR studies require more subjects, longer durations, complex statistical methods like RSABE, and extensive dissolution testing across multiple pH levels. The clinical trials themselves cost 30-50% more than IR studies, and failure rates are higher-about 22% of MR applications are initially rejected by the FDA.
What’s the future of modified-release bioequivalence?
The future is moving toward modeling and reduced human testing. In vitro-in vivo correlation (IVIVC) models are being accepted by the FDA to predict how a drug will behave in the body based on lab dissolution data. Physiologically based pharmacokinetic (PBPK) models are also becoming common, allowing developers to simulate absorption under different conditions without running full clinical trials. These tools could cut development time and cost-but they require deep expertise and high-quality data. For now, they’re mostly used by large pharma companies, but they’re the key to making complex generics more accessible.